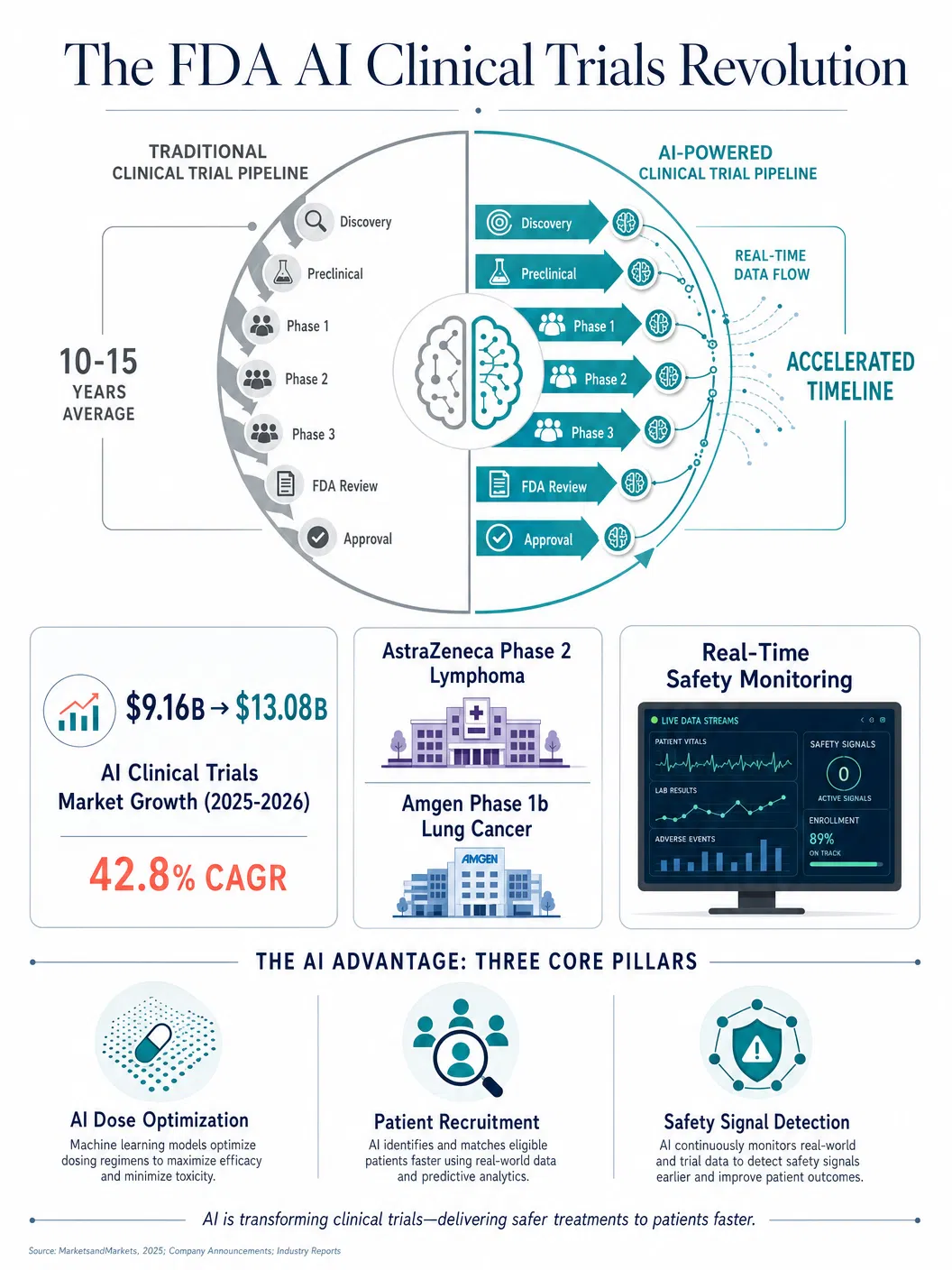
On April 28, 2026, the United States Food and Drug Administration announced an initiative that may prove to be one of the most consequential regulatory decisions in modern pharmaceutical history. Under the leadership of Commissioner Marty Makary, the agency launched a pilot programme to review clinical trial data in real time, beginning with trials conducted by two of the world's largest pharmaceutical companies: AstraZeneca and Amgen. Simultaneously, the FDA issued a public request for information on a broader pilot programme that would integrate artificial intelligence into safety monitoring, medication dose selection, safety signal identification, and patient recruitment across clinical trials. Together, these actions signal a fundamental shift in how the world's most influential drug regulator approaches the evaluation of new medicines.
The traditional model of clinical trial oversight operates on a sequential, retrospective basis. Pharmaceutical companies conduct trials, collect data over months or years, compile that data into massive regulatory submissions, and then wait for the FDA to review the package — a process that itself can take months to a year or more. During this time, patients who might benefit from effective treatments continue to wait, and safety signals that emerge during the trial may not be fully analysed until long after they first appear. The cumulative effect is a drug development timeline that averages ten to fifteen years from initial discovery to market approval, with costs frequently exceeding two billion dollars per approved drug.
The FDA's new pilot programme attacks this inefficiency at its structural core. AstraZeneca's participation involves a Phase 2 trial of a combination therapy for patients with an aggressive form of lymphoma, conducted at the University of Texas MD Anderson Cancer Center and the University of Pennsylvania — two of the nation's premier cancer research institutions. Amgen is contributing a Phase 1b trial of a treatment for small cell lung carcinoma. Both trials will utilise a real-time data platform built by Paradigm Health, which enables the FDA to access and analyse trial data as it is generated rather than waiting for periodic reports or final submissions.
The implications of real-time data review extend far beyond administrative convenience. When regulators can observe trial data as it accumulates, they can identify safety concerns earlier, potentially protecting participants from adverse events that might otherwise go undetected for months. They can also recognise efficacy signals sooner, enabling faster decisions about whether to expand trials, modify dosing protocols, or accelerate the path to approval. For oncology trials in particular, where patients often have limited treatment options and rapidly progressing disease, the difference between real-time and retrospective review can be measured in lives.
The AI component of the FDA's initiative addresses an equally fundamental challenge: the sheer volume and complexity of modern clinical trial data. Contemporary trials generate enormous datasets encompassing genomic profiles, biomarker measurements, imaging data, patient-reported outcomes, electronic health records, and real-world evidence from wearable devices. No human review team, regardless of its expertise, can process this information in real time. AI systems, by contrast, can continuously monitor incoming data streams for patterns that indicate safety signals, identify optimal dosing ranges based on pharmacokinetic modelling, and even predict which patient subpopulations are most likely to benefit from a given therapy.
The market context underscores the urgency of this transformation. The AI in clinical trials market reached an estimated 9.16 billion dollars in 2025 and is projected to grow to 13.08 billion dollars in 2026, reflecting a compound annual growth rate of 42.8 percent. This explosive growth is driven not by speculative investment but by demonstrated utility: AI-powered patient recruitment tools have been shown to reduce enrolment timelines by thirty to fifty percent in some studies, while machine learning algorithms for safety monitoring can detect adverse event patterns that traditional statistical methods miss entirely.
The regulatory philosophy underlying the FDA's initiative represents a departure from the agency's historically conservative approach to new technologies. Commissioner Makary, who took office with a mandate to modernise the FDA's operations, has framed the pilot programme as part of a broader effort to make the agency more responsive to technological innovation without compromising its core mission of protecting public safety. The request for public information on AI integration suggests that the agency is approaching this transformation methodically, seeking input from industry, academia, and patient advocacy groups before establishing permanent regulatory frameworks.
Critics of the initiative raise legitimate concerns. Real-time data access creates new challenges around data integrity, patient privacy, and the potential for premature regulatory decisions based on incomplete datasets. There is also the question of whether AI systems, which are trained on historical trial data, can reliably identify novel safety signals that fall outside their training distribution — a concern that echoes broader debates about AI reliability in high-stakes decision-making. The pharmaceutical industry itself has mixed incentives: while faster approvals benefit companies with promising drugs, real-time scrutiny could also lead to earlier termination of trials that might otherwise have continued long enough to show marginal benefits.
The international implications are equally significant. If the FDA's pilot programme demonstrates that real-time AI-assisted trial review can safely accelerate drug development, regulatory agencies worldwide will face pressure to adopt similar approaches. The European Medicines Agency, Japan's Pharmaceuticals and Medical Devices Agency, and China's National Medical Products Administration are all watching the FDA's experiment closely. The agency that sets the standard for AI-integrated drug regulation will shape the global pharmaceutical landscape for decades to come.
What makes the FDA's 2026 initiative genuinely transformative is not any single technological capability but the institutional willingness to reimagine the fundamental relationship between regulators and the data they evaluate. For seventy years, that relationship has been defined by distance — temporal, analytical, and procedural distance between the generation of clinical evidence and the regulatory decisions based upon it. The real-time AI pilot programme collapses that distance, creating the possibility of a regulatory process that is not merely faster but fundamentally more responsive to the biological realities of disease and treatment. Whether this experiment succeeds will depend on execution, but the ambition alone marks a turning point in the history of drug regulation.