The sequencing of the human genome at the turn of the millennium was celebrated as a landmark achievement — and rightly so. But in retrospect, it was less a destination than a starting point. The genome sequence itself, a string of three billion nucleotide letters, is largely uninterpretable without the analytical tools to identify which variations matter, which genes are active in which cells, and how the proteins encoded by those genes fold into three-dimensional structures that determine their function. For two decades, progress on all three fronts was constrained by the limitations of classical statistical and computational methods. Machine learning is now lifting those constraints, enabling analyses of a depth and scale that were previously impossible.
SNP Analysis: Finding the Variants That Matter
Single nucleotide polymorphisms — SNPs, pronounced "snips" — are the most common form of genetic variation in the human genome, occurring at a frequency of roughly one per thousand base pairs. The human genome contains approximately 4 to 5 million SNPs per individual, and genome-wide association studies (GWAS) have identified thousands of SNPs associated with complex diseases including type 2 diabetes, schizophrenia, coronary artery disease, and dozens of cancers. The challenge is not finding SNPs — modern sequencing platforms can identify them comprehensively — but interpreting which ones are functionally significant and how they contribute to disease risk.
Classical GWAS approaches identify statistical associations between SNPs and phenotypes, but they cannot distinguish causal variants from those that are merely in linkage disequilibrium with the true causal site. They also struggle with rare variants, gene-gene interactions, and the interpretation of non-coding variants, which constitute the vast majority of GWAS hits but whose functional consequences are difficult to predict from sequence alone.
Machine learning is addressing each of these limitations. Deep learning models trained on chromatin accessibility data, histone modification profiles, and transcription factor binding data can predict whether a non-coding SNP disrupts a regulatory element — providing a mechanistic hypothesis for GWAS associations that previously had no clear functional interpretation. Models such as DeepSEA, Basenji, and Enformer take raw DNA sequence as input and predict hundreds of epigenomic features simultaneously, enabling researchers to score the regulatory impact of any SNP in any genomic context.
| ML Model | Input | Output | Key Application |
|---|---|---|---|
| DeepSEA | 1kb DNA sequence | 919 chromatin features | Prioritising non-coding GWAS variants |
| Enformer | 196kb DNA sequence | Gene expression predictions | Long-range regulatory variant effects |
| CADD | Sequence + annotations | Pathogenicity score | Clinical variant interpretation |
| AlphaMissense | Protein sequence context | Missense variant pathogenicity | Protein-coding SNP interpretation |
| PolyPhen-2 | Amino acid sequence | Functional impact score | Coding variant annotation |
| SpliceAI | Pre-mRNA sequence | Splicing disruption score | Splice-site variant interpretation |
For population genomics and biosecurity applications — including the identification of SNPs associated with antimicrobial resistance in bacterial pathogens, or the tracking of viral evolution during outbreaks — machine learning models trained on pathogen genome databases can identify resistance-conferring mutations with sensitivity and specificity that exceed classical rule-based approaches. This has direct implications for the design of diagnostic assays, the selection of therapeutic targets, and the real-time genomic surveillance of emerging infectious diseases.
Single-Cell RNA-Seq: Resolving Cellular Heterogeneity
Single-cell RNA sequencing (scRNA-seq) has transformed our understanding of cellular biology by enabling the transcriptional profiling of individual cells rather than bulk tissue averages. A single scRNA-seq experiment can generate expression profiles for tens of thousands of cells simultaneously, revealing the cellular composition of complex tissues, identifying rare cell populations, and tracing the developmental trajectories of differentiating cells. The technology has been applied to map the cellular atlases of the human brain, lung, liver, and immune system, and to characterise the tumour microenvironment in unprecedented detail.
The analytical challenge posed by scRNA-seq data is formidable. A typical experiment generates a matrix of gene expression values for 20,000 genes across 10,000 to 100,000 cells — a high-dimensional, sparse, and noisy dataset that defies classical statistical approaches. The key analytical tasks — dimensionality reduction, cell type annotation, trajectory inference, differential expression analysis, and batch effect correction — all benefit enormously from machine learning.
Variational autoencoders (VAEs) such as scVI and its successors have become the workhorses of scRNA-seq analysis, learning low-dimensional latent representations of cellular transcriptomes that capture the major axes of biological variation while accounting for technical noise and batch effects. Graph neural networks are being used to model the spatial relationships between cells in tissue sections, integrating transcriptomic and spatial information to reconstruct the cellular architecture of organs. Transformer-based models — including the Geneformer and scGPT foundation models, trained on millions of single-cell transcriptomes — can predict the transcriptional consequences of gene perturbations, enabling in silico drug target identification without the need for expensive experimental screens.
For infectious disease research, scRNA-seq combined with machine learning has revealed how individual immune cells respond to pathogen challenge — identifying the transcriptional signatures of protective versus pathological immune responses, and pointing toward new targets for vaccine and therapeutic development. In the context of biosafety, these tools are enabling a more mechanistic understanding of how pathogens manipulate host cell biology, with implications for the design of countermeasures against both natural and engineered biological threats.
Protein Structure and Function: The AlphaFold Revolution and Beyond
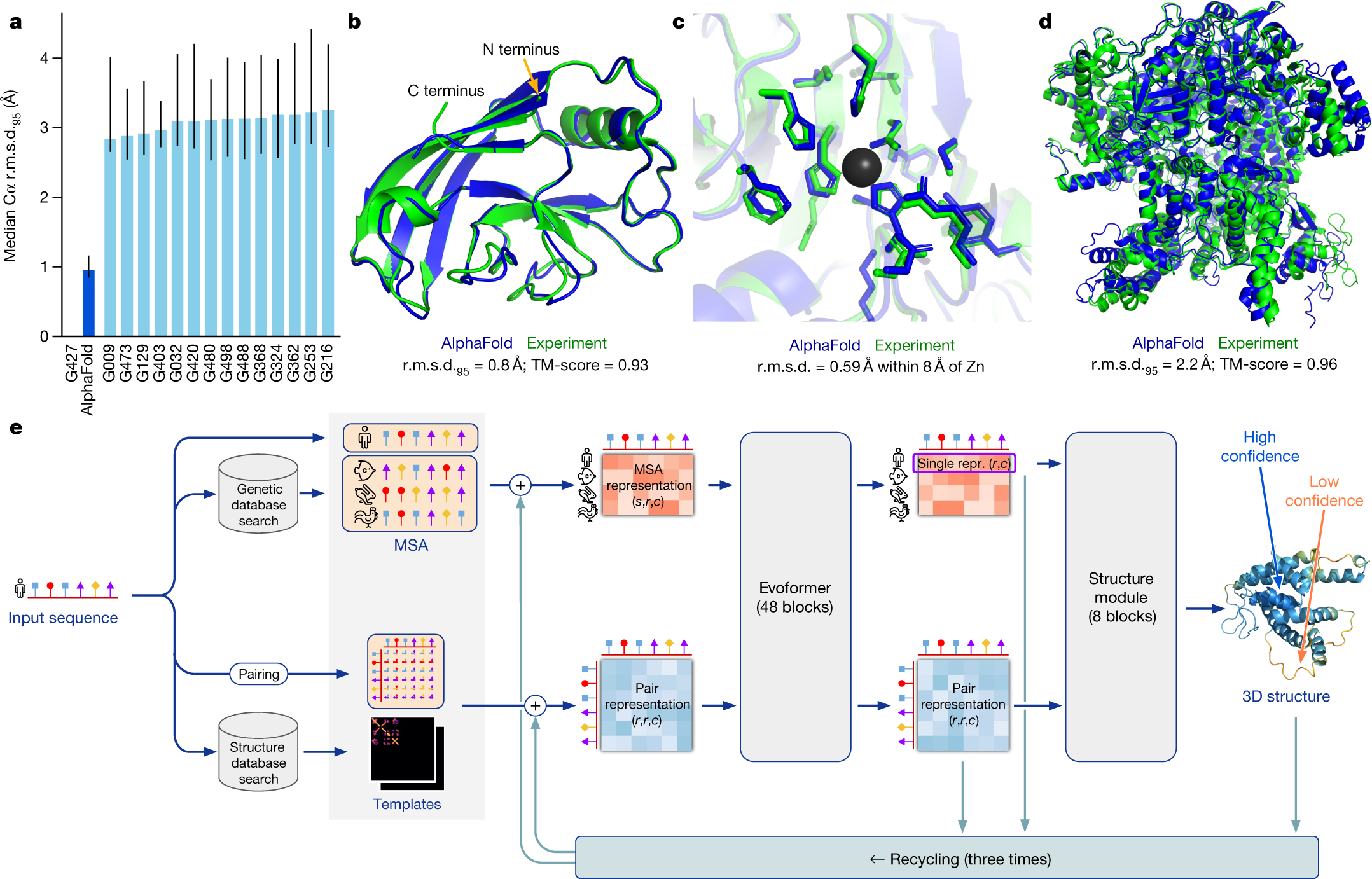
The prediction of protein three-dimensional structure from amino acid sequence — the protein folding problem — was for fifty years one of the grand unsolved challenges of biology. The solution, when it came, was delivered by a deep learning system. DeepMind's AlphaFold2, released in 2021, achieved a level of structural prediction accuracy that matched experimental methods for the majority of protein families, effectively solving the protein folding problem for most practical purposes. The AlphaFold Protein Structure Database now contains predicted structures for over 200 million proteins — essentially the entire known protein universe.
The implications for biosafety, drug discovery, and biotechnology are profound. Researchers can now obtain high-quality structural models for any protein of interest within minutes, enabling structure-based drug design, the identification of binding sites for small molecules, and the engineering of proteins with novel functions. For biosecurity applications, AlphaFold has been used to predict the structures of proteins encoded by poorly characterised pathogens, providing structural hypotheses for the mechanisms of virulence and drug resistance that can guide experimental validation.
Beyond structure prediction, machine learning is being applied to the harder problem of function prediction — determining what a protein does from its sequence and structure. Language models trained on protein sequences, such as ESM-2 and ProteinBERT, learn representations that capture functional relationships between proteins, enabling zero-shot prediction of enzyme activity, binding specificity, and subcellular localisation. These models are being used to annotate the vast dark matter of the protein universe — the millions of proteins of unknown function that populate microbial genomes — and to design novel proteins with specified functional properties.
The convergence of structural prediction, function annotation, and generative protein design is creating a new paradigm for protein engineering. Rather than iterating through random mutagenesis libraries, researchers can now design proteins computationally, predict their structure and function with high confidence, and synthesise only the most promising candidates for experimental validation. This is accelerating the development of new enzymes for industrial biotechnology, new antibodies for therapeutic applications, and new biosensors for environmental monitoring — all with direct relevance to the biosafety and biosecurity challenges of the coming decades.